? 27 min
Što je kliničko istraživanje i kako (i zašto) funkcionira?
Kliničko istraživanje je studija na ljudima – dobrovoljcima, kako bi se pronašli odgovori na pitanja vezana za određenu terapiju/intervenciju.
- je li terapija/intervencija sigurna?
- da li terapija/intervencija funckionira?
- funckionira li terapija/intervencija bolje od one koja je već dostupna (ukoliko takva terapija postoji)?
Klinička istraživanja moraju biti provedena u skladu sa međunarodnom konferencijom za harmonizaciju (engl. International Conference on Harmonization (ICH)) i dobrom kliničkom praksom (engl. Good Clinical Practice (GCP)) i moraju biti odobrena prije početka od strane više regulatornih ili kompetentnih Agencija (eng. Regulatory or Competent Authorities (CAs)) i etičkih povjerenstava (eng. Ethics Committees (ECs)), ovisno o državi u kojoj bi se studija provodila. Takve regulatorne agencije uključuju Agenciju za hranu i lijekove Sjedinjenih Američkih Država – engl. Food and Drug Administration; FDA, Europsku agenciju za lijekove – engl. European Medical Agency; EMA (i dodatna razna regulatorna tijela u zasebnim državama Europske Unije), Regulatornu agencija za lijekove i zdravstvene proizvode -engl. Medicines and Healthcare products Regulatory Agency; MHRA u Ujedinjenom Kraljevstvu i Agenciju za lijekove i medicinske proizvode – engl. Pharmaceuticals and Medical Devices Agency; PMDA u Japanu. Primjeri etičkih povjerenstava su Institutional Review Board (IRB) u Sjedinjenim Američkim Državama i Medical Research Ethics Committee (MREC) u Nizozemskoj. Ova tijela razmatraju planove za provedbu kliničkih istraživanja i bez njihovog odobrenja i sporazuma, studija ne može započeti.
Komponente kliničkih istraživanja
Kako bi se kliničko istraživanje provelo, potrebao je ustanoviti precizan i detaljan plan provedbe. Također, potrebno je identificirati cijeli niz bitnih stranaka povezanih sa studijom, te razviti temeljit i pedantan logistički i znanstveni plan u skladu s dizajnom studije i samom provedbom.
Protokol i informirani pristanak
Najbitniji dokument u kliničkim istraživanjima je protokol studije, u kojem su sadržani dizajn studije, ciljevi istraživanja i parametri koji se mjere, metode, procjene i testiranja, raspored aktivnosti i procedura i statističke stavke bitne za analizu podataka. Protokol također opisuje korake bitne za zaštitu prava sudionika i način na koji studija procjenjuje sigurnost sudionika uključujući i sve kriterije uključivanja i isključivanja, kojih se sudionik mora 100% pridržavati. Primjerice, ako kriterij za uključivanje sudionika u studiju kaže da sudionik mora imti više of 18 godina, ukoliko ima 17 godina i 11 mjeseci, ne smije biti uključen. Nepoštivanje kriterija uključivanja i isključivanja sudionika može dovesti do ozbiljnih posljedica – ne samo ugroziti studiju i novu terapiju kao takvu, nego ugroziti i ugled svih uključenih, te u najgorem slučaju dovesti do ozbiljnih zdravstvenih ishoda.
Još jedan važan dokument u kliničkim istraživanjima, namjenjen sudionicima – dobrovoljcima je obrazac za informirani pristanak (engl. Informed Consent Form (ICF)) koji opisuje studiju na laički način i sadrži načine procjene sigurnosti, prava sudionika i što se točno događa s uzorcima i podatcima prikupljenima u studiji. Ukoliko sudionik nije podoban za sudjelovanje u studiji ili ukoliko ne razumije istraživanje, što sudjelovanje u studiji znači te ne da svoj pristanak, takav sudionik ne može pristupiti studiji.
Nasumično kontrolirana ispitivanja/randomizirane kontrolirane studije
Kada raspravljamo o kliničim istraživanjima koja se bave ispitivanjem lijekova ili cjepiva, iako postoje i drugi oblici istraživanja, najčešće je to u kontekstu nasumično kontroliranih ispitivanja/randomiziranih kontroliranih studija (engl. randomized controlled trials (RCTs)). Takve studije predstavljaju zlatni standard u kliničkim ispitivanjima. One su nasumične jer ukoliko gledamo podobne sudionike:
a) uzorak iz populacije koji će biti izložen novoj terapiji/cjepivu je izabran nasumično, te
b) uzorak iz populacije koji neće biti izložen novoj terapiji/cjepivu (ili preciznije) koji će biti izložen placebu/sličnoj terapiji je također izabran nasumično.
Takva studija, kada imamo jednu grupu koja je izložena intervenciji (u stvari prima intervenciju) a druga grupa je izložena placebu ili sličnoj intervenciji (komparatoru), je kontrolirana studija. Koristeći kontrolirani dizajn, namjera je ustanoviti da je djelovanje lijeka koji vidimo istinit a ne slučajan.
Placebo (također znan i kao “šećerna pilula”) je najčešće lijek ili cjepivo koje izgleda potpuno jednako i koji se upotrebljava na potpuno jednak način kao i lijek/cjepivo s aktivnim sastojcima. Ponekad korištenje placeba u kliničkim istraživanjima nije poželjno, i umjesto njega, drugi komparator lijek/cjepivo s aktivnim sastojcima, od kojeg sudionici imaju dokazane koristi, se može upotrijebiti. Usporedba između grupa koje koriste placebo/komparator i aktivnu intervenciju se često provodi na slijepi način, tako da ni sudionici ni istraživači ne znaju tko je primio koju terapiju sve dok se rezultati ne analiziraju i dok se “kod ne razbije”.
Pojedinci u studiji bi trebali imati slične osnovne osobine tako da je jedina varijacija izlaganje intervenciji (lijeku, cjepivu, uređaju, procesu) koja se istražuje. Slične osnovne osobine među grupama su nužne kako bi se smanjila bilo kakva pristranost prilikom interpretacije rezultata. Proces randomizacije ili nasumičnosti (raspodjela svakog pojedinca u grupu studije na nasumičan način) osigurava da su sudionici raspodijeljeni u grupe nasumično, time osiguravajući ravnotežu između grupa prema osnovnim osobinama. Takav proces se koristi kako bi se smanjila pristranost u odabiru (engl. selection bias). Postoji cijeli niz važnih osnovnih osobina, no one koje se najčešće koriste su spol, dob, rasa i druge za studiju bitne karakteristike (npr. prisutstvo/odsutstvo biomarkera važih za bolest). Strategije randomizacije koje se često upotrebljavaju su stratifikacija i minimizacija. Stratificirana randomizacija ili stratifikacija se može koristiti kada se grupe u studiji žele dodatno podijeliti u blokove (strate) prema faktoru koji nije intervencija (npr. prema spolu = muškarci vs. žene unutar grupa), pritom pazeći da su blokovi uravnoteženi/homogeni. Stratifikacija se često koristi ukoliko u studiji postoji plan za dodatne analize prema podgrupama (npr. prema spolu). Minimizacija, nasuprot stratifikaciji, je više fleksibilna i dopušta dimaničnu/prilagodljivu randomizaciju kako bi smanjila neuravnoteženost među grupama, uzimajući u obzir “zbunjujuće čimbenike” (engl. confounding factors). Zbunjujući čimbenk je varijabla koja ima utjecaj na neovisnu i ovisnu varijablu. Tako npr., dob može biti zbunjujući čimbenik:
- Za razvoj/pogoršanje bolesti dob može biti važna jer može pridonijeti pogoršanju, pri čemu je bolest ovisna varijabla = ishod koji mjerimo u studiji (razvitak bolesti ili pogoršanje ovisno o dobi).
- Dob također može imati utjecaja na učinkovitost terapije za određenu bolest, pri čemu je terapija neovisna varijabla = terapija koja se razlikuje među grupama u studiji (određena terapija može bolje djelovati na mlađu populaciju, npr.).
I dok je stratifikacija često predodređena, minimizacija se prilagođava regrutiranju sudionika i radi na način da smanjuje neuravnoteženosti, što može biti korisno kada je uzorak sudionika mali. Obje metode imaju svoje prednosti i nedostatke, te razlike u upotrebi u kliničkim istraživanjima ovisno o ciljevima studije.
Ciljevi istraživanja i parametri kliničkih studija, te statističko testiranje (hipoteze)
Parametri (ili ishodi) istraživanja, određenih za svakog sudionika studije, su kvantitativne mjere zadane prema ciljevima istraživanja. Kao takvi, parametri mogu biti, npr.:
- broj nuspojava u grupi koja prima terapiju nasuprot istom u placebo grupi tijekom jedne godine, ili
- preživljavanje nakon 5 godina pod novom terapijom u onkološkoj studiji na pacijentima u terminalnoj fazi karcinoma.
Kliničke studije u pravilu imaju glavni cilj i odgovarajauće glavne parametre prema kojima se određuje početni broj sudionika = veličina uzorka u studiji. Dodatni ciljevi i parametri su sporedni ili sekundarni i oni se moraju zasnivati na veličini uzorka određenim prema glavnom cilju. Osim njih, postoje još i tercijarni ili istraživački ciljevi i parametri koji ne utječu na veličinu uzorka ali dodatno podržavaju glavne i sekundarne ciljeve.
Parametri koji se koriste u kliničkim istraživanjima moraju biti u skladu sa znanstvenim ciljevima studije i metode koje istražuju ishode moraju biti što je više moguće, nepristrane i točne. Neki primjeri parametara su:
- Kontinuirana (neprekinuta) mjerenja: krvni pritisak, težina – tijekom vremenskog perioda.
- Vrijeme događaja: vrijeme do povrtaka karcinoma, vrijeme preživljenja – dok se događaj ne pojavi.
- Uređene kategorije: odsutna, blaga, snažna bol.
Da bi se ustanovili klinički ishodi i parametri, važno je definirati koje pretpostavke/hipoteze želimo istražiti. U standardnom, tipičnom kliničkom istraživanju to će značiti istraživanje superiornosti jedne grrupe (obično one koje prima novi lijek) nasuprot grupe koja prima kontrolu/komparator (može biti placebo ili druga terapija s aktivnim sastojcima). Ono što će se proučavati jest da li je jedna grupa bolja od druge u sličnoj grupi ljudi za jedan ili više prethodno određenih parametara.
Kako bi se ustanovilo je li novi lijek bolji od komparatora ili placeba na nepristran, statistički ustanovljen način, testiranje hipoteze se primjenjuje prilikom analize studije, za koji se unaprijed (prije nego studija započne) definira nulta i alternativna hipoteza:
Nulta hipoteza (H0) kliničih studija (superiornosti) tvrdi da nema prave/istinite razlike između dviju intervencija (kontrolne i aktivne grupe).
Alternatina (druga) hipoteza (H1) je suprotna od H0 i tvrdi da postoji razlika između dvije intervencije za glavni cilj studije.
Za uspostavljanje ishoda da je jedan lijek bolji (superioran) sa statističkom sigurnošću, moramo moći odbaciti nultu hipotezu. To se najčešće čini računanjem p-vrijednosti, koja se derivira raznim statističkim testovima, ovisno o tipu studije.
Pod pretpostavkom da je nulta hipoteza točna, p-vrijednost je broj koji opisuje koliko je vjerojatno da su podatci koje vidimo slučajni. P-vrijednost je broj između 0 i 1 i što je manji, veća je mogućnost da se nulta hipoteza odbaci. Ako je p-vrijednost manja od razine značajnosti koju smo definirali, odbacit ćemo nultu hipotezu. Obično, razina značajnosti je unaprijed definirana kao 0.05 i ako je p-vrijednost manja od 0.05, ona podupire snažan dokaz protiv nulte hipoteze, jer postoji manje od 5% vjerojatnosti da je H0 istinita (i da su rezultati koje vidimo slučajni). Zato, odbacujemo nultu hipotezu i prihvaćamo alternativnu hipotezu koja kaže da su dvije terapije različite.
Prilikom analize rezultata kliničkih istraživanja i prilikom izvršavanja statističkog testiranja, postoji rizik da se donesu pogrešni zaključci, time ugrožavajući zaključke same studije. Rizici se mogu smanjiti pomnim planiranjem studije, a najprije opreznim računanjem potrebnog broja sudionika, tzv. ispravne veličine uzorka ne bi li se dali ispravni odgovori na pitanja studije i odabirom bitnih glavnih parametara. Upotrebom prikladnih statističkih metoda za računanje primjerene veličine uzorka za rješavanje istraživačkih pitanja, pogreške se mogu reducirati.
Dvije najčešće pogreške su Pogreška vrste I = lažno pozitivan zaključak koji ukazuje da su dvije terapije različite, dok u stvarnosti nisu (engl. Type I error = a false positive), i Pogreška vrste II = lažno negativan zaključak koji ukazuje da dva tretmana nisu ražličita, iako u stvarnosti jesu (engl. Type II error = a false negative). Pogreška vrste I je ozbiljna i može ugroziti ne samo jednu studiju nego predstaviti i rizik za ugled te stvoriti nenaklonost prema farmacutskim proizvodima. Ona bi značila da vidimo učinkovitost novog lijeka ili da je novi lijek bolji od starog, kada u stvarnosti to nije tako.
| Stvarnost | |||
| Istina | Laž | ||
| Izmjereno ili zamijećeno (u studiji) | Istina | Ispravna odluka (stvarno pozitivan) | Pogreška vrste I (lažno pozitivan) |
| Laž | Pogreška vrste II (lažno negativan) | Ispravna odluka (stvarno negativan) |
Tijekom analize, kada je studija završena, istraživači moraju izabrati kako će pristupiti prikupljenim podatcima. Podatci koji se skupljaju mogu biti nuspojave, uzorci krvi, krvni pritisak, rentgenske snimke, itd., prije i nakon nakon nove i kontrolne terapije. Pristup namjere-za-liječenjem (engl. Intent-To-Treat (ITT) – svi koji su uključeni u studiju i za koje postoji namjera da se izlože inervenciji), koji uzima sve sudionike randomizirane u studiji, bez izostavljanja, se obično koristi u randomiziranim kontroliranim studijama. Takva analiza ne izostavlja bilo kakva odstupanja od protokola, odnosno podatke koji jasno krše pravila definirana u protokolu, no zbog toga više nalikuje situaciji u stvarnom svijetu. Ne uzima u obzir prijevremeni odlazak sudionika iz studije, odbijanje terapije ili nepoštovanje pravila za analizu. Na primjer, ako imamo sudionika koji je trebao dati krvni uzorak 30 dana nakon cijepljenja u studiji, a na kraju je krvi uzorak uzet tek 60 dana nakon cijepljenja, pristup namjere-za-liječenjem bi svejedno uključio ovog sudionika u analizi, iako interval koji je definiran prema protokolu nije ispoštovan. Nasuprot, analiza prema-protokolu (engl. Per-Protocol Set (PPS)) donosi zaključke na podatcima koju su strogo vjerni standardima definiranima u protokolu i time je više kontrolirana. U prethodnom primjeru, sudionik kojem je krvni uzorak uzet 60 dana nakon cijepljenja će biti izuzet iz analize prema-protokolu. Općenito, najbolji i preporučeni pristup je izvršiti obje analize ITT i PPS, i ako obje dovedu do istih zaključaka, studija i rezultati se smatraju vrlo pouzdanima.
Naposljetku, kliničko istraživanje može imati više analiza tijekom studije. Ponekad, posebno ako studija traje dugo i/ili je dizajn kompliciran/složen prilikom čega jedan dio može ovisiti o rezultatima drugog dijela, privremene analize (engl. interim analyses) se mogu provesti kako bi se procijenili trenutni podatci u studiji prije nego što se izvrši završna analiza (engl. final analysis) koja sadrži podatke iz cijele studije.
Zasljepljivanje/maskiranje kliničkih istraživanja
Zasljepljivanje (engl. blinding) ili maskiranje je važan element kliničkih istraživanja, prilikom čega sve ili neke strane povezane sa studijom ne znaju koji lijek/cjepivo je svaki sudionik primio. U slijepim studijama, terapije su maskirane tako da izgledaju isto (npr. cjepivo i placebo su jednako pakirani u identične injekcije tako da ih nitko ne može razlikovati).
Dvostruko-slijepa (engl. double-blind) studija je ona u kojoj su i sudionik koji prima terapiju i osoba koja daje lijek/cjepivo slijepe, te ne znaju koji lijek/cjepivo sudionik prima. Jednostuko-slijepa (engl. single-blind) studija, pri kojoj je samo jedna strana (ili sudionici ili istraživači), slijepa, se također često provodi, kao i otvorene studije (engl. open-label) u kojima nema maskiranja.
Maskiranje se koristi kako bi se spriječila pristranost kako onih koji rade u klinikama gdje se studija provodi, tako i samih sudionika. Ako se maskiranje ne koristi, postoji šansa da sudionici budu pristrani prilikom prijave nuspojava, posebno ukoliko postoji sklonost ka jednoj terapiji. Štoviše, ako je jedna od terapija u studiji placebo, maskiranje osigurava da nema efekta placeba, t.j. ako jedan sudionik zna da prima cjepivo s aktivnim sastojkom, a drugi prima placebo, mogla bi postojati pristranost prilikom prijave nuspojava – snažnija bol prijavljena nakon aktivnog cjepiva naspram blaže boli prijavljene nakon placeba, dok je u stvarnosti bol slična.
Ponekad upotreba placeba i maskiranja nije etična. Kada postoji dokazana korist novog lijeka s prikladnim sigurnosnim profilom i minimalnim nuspojavama, i ne postoji alternativni lijek, svako sljedeće istraživanje mora biti otvorenog tipa jer uporaba placeba nije primjerena i nema drugog lijeka koji se može dati bolesnicima. Također, ako su sudionici pacijenti koji ovise o terapiji, nije etično dati im placebo – u takvim slučajevima jedna grupa koristi novi lijek dok druga prima standardnu terapiju za istu bolest.
Važne uloge u kliničkim istraživanjima
Klinička studija je izrazito složena platforma i iziskuje sudjelovanje više raznih stranaka. Osim toga, studije su iznimno regulirane i postoje definirane obaveze o pravovemenom i trajnom izvještavanju o podatcima i rezultatima prema regulatorima, etičkim povjerenstvima, znanstvenim časopisima, itd. U osnovi, da bi se nova terapija razvila, potrebno je ostvariti više raznih partnerstva koja su ponekad definirana posebnim ugovorima, ovisno o odnosu.
Pokretač studije je općenito sponzor, npr. farmaceutska tvrtka ili istraživačka bolnica/institut zainteresirana za istraživanje nove terapije/lijeka/cjepiva/uređaja = u osnovi novog proizvoda ili novih postupaka. Uz sam razvoj proizvoda, sponzor obično dizajnira studiju, protokol i odgovarajuće ciljeve te potrebe studije (kao što je broj sudionika, njihove obaveze i koristi vezane za sudjelovanje u studiji), plan koji opisuje kako se podatci prikupljaju i analiziraju itd. Oni također predlažu države u kojima bi se studija trebala provesti (npr., izbor između sjeverne i južne polutke ako je to važno za primjenu sezonskih cjepiva: obično se cjepivo, primjerice protiv gripe, daje u zimskim mjesecima koji se razlikuju na sjeveru i jugu) i procjenjuju klinke/lokacije koje bi mogle biti uključene. Sponzor isto može biti i glavni ulagač, no ulagači mogu biti i neprofitne organizacije kao što je The Bill & Melinda Gates Foundation.
Osoblje koje provodi studiju, koje ima izravni doticaj sa sudionicima i koje prikuplja podatke su obično bolnice/klinike s prethodnim iskustvom u provođenju kliničkih studija. Takvo osoblje obično čine medicinske sestre, koordinatori, sub-istraživači i glavni istraživači studije. Takav tim pri istraživačkoj lokaciji je najčešće pripremljen za provodbu kliničkih studija i spreman da se aktivira u bilo kojem trenutku. Izbor lokacije će ovisiti o tome treba li nužno uključiti određenu državu zbog posebnog razloga ali će također ovisiti o populaciji koja se treba proučavati: djeca, starija populacija, trudnice ili pacijenti. To znači da lokacija/tim mora posjedovati stručnost u određenoj grani medicine, ovisno o bolesti/lijeku koje se istražuju.
Jedna od najvažnijih stranaka u kliničkim studijama je sudionik, zdravi dobrovoljac/dobrovoljka ili pacijent/pacijentica koji može i želi sujelovati u konkretnom istraživanju. Bez sudionika, kliničke studije ne bi postojale i naposljetku, ne bi bilo unapređenja kliničke njege i terapija. To znači da je iznimno važno stvoriti i zadržati povjerenje, transparentnost i poštovanje prije, tijekom i nakon studije sa sudionikom. Važna značajka sudjelovanja u kliničkim studijama je prikupljanje podataka/uzoraka, analiza i dijeljenje i prirodno je da postoji rastuća zabrinutost vezana za zaštitu podataka. Danas su podatci (uključujući uzorke) anonimizirani i kodirani, te su dodatno zaštićeni na najvišoj mogućoj razini. Također, sudionik obično ima pravo zatražiti da odustane od sudjelovanja u studiji i u skladu s time da se svi podatci i uzorci prikupljeni unište u bilo kojem trenutku.
Etička povjerenstva, kao što je IRB u Sjedinjenim Američkim Državama, imaju važnu ulogu u kliničkim istraživanjima. Njihov glavni zadatak je da osiguraju da su studije etične i da su sudionici u studiji poštovani i zaštićeni. Njihova uloga je pregledavanje dokumenata istraživanja, revizija ukoliko je potrebna i odobrenje ili odbijanje studije, pri čemu je interes sudionika najbitnija stavka. Jedan od najvažnijih dokumenata kojima se etička povjerenstva bave (jednako važan i za sudionika) je obrazac za informirani pristanak.
Uključujući sve gore-navedene stranke koje odobravaju studiju, studija ne može započeti prije nego je odobrena od strane Regulatornih ili Kompetentnih Agencija. Agencija za hranu i lijekove Sjedinjenih Američkih Država – engl. Food and Drug Administration; FDA, Europska agencija za lijekove – engl. European Medical Agency; EMA su primjeri takvih regulatornih tijela. Svaka država također ima svoje zdravstvene agencije ili tijela i pridružene pravilnike (odredbe i smjernice) te normative kojih se studija, za koju sponzor traži odobrenje, mora pridržavati. Slično etičkim povjerenstvima, zdravstvene agencije pregledavaju, vrše reviziju, te i odobravaju ili odbijaju studiju, ali ne fokusirajući se samo na sudionika nego na cijelu studiju, uključujući i znanstvene i logističke značajke. Klinička istraživanja su regulirana kako bi poduprla ravoj medicinskih proizvoda, osiguravajući pritom istovremeno da će rezultati koji nastaju služiti svojoj svrsi – a to je dokaz da je terapija sigurna i djelotvorna.
Odbor za praćenje sigurnosti podataka (engl. Data Safety Monitoring Board (DSMB) i/ili Data Monitoring Committee (DMC)) obično služe kao vanjsko tijelo koje pruža trajni i kontinuirani nadzor nad prikupljenim podatcima o sigurnosti i savjetuje bi li studija trebala nastaviti ili se zaustaviti u bilo kojem vremenu ukoliko su rizici utvrđeni. Takav odbor čini grupa stručnjaka – obično liječnika, znanstvenika i statističara odgovornih za pregled nemaskiranih podataka i praćenje kvalitete te procjena omjera rizika/koristi za svakog sudionika i za sve sudionike zajedno. Ako studija ima pravila zaustavljanja/stopiranja (engl. holding/stopping rules – set pravila koji vode do zaustavljanja studije kada se ustanovi rizik) odbor za praćenje sigurnosti podataka može zaustaviti studiju.
Kako su klinička ispitivanja složena i kako podliježu regulacijama koje se neprestano mijenjaju, sponzori obično angažiraju vanjske suradnike i usluge koji pomažu s provedbom studije. Razni dobavljača mogu sudjelovati u studiji, kako bi pomogli s laboratorijskim testiranjem i procesiranjem, pošiljkama, dnevnicima sudionika (papirnati ili elektronički dnevnici preko kojih se nuspojave prijavljuju), reklamiranjem i regrutiranjem, itd.
Naposljetku, kada je studija gotova i podatci analizirani, rezultati se moraju objaviti i javno obznaniti u prethodno određenom vremenskom periodu od zadnjeg posjeta sudionika u studiji. Takva obaveza ne postoji samo prema regulatorima, sudionicima u studiji i osoblju koje radi na istraživanju, nego i prema široj javnosti. Izuzev dijeljenja rezultata kao obaveze, obično se rezultati također objave u stručnim časopisima i na kongresima i konferencijama.
Rezultati više studija se koriste u pripremi zahtjeva za podnošenje regulatornom tijelu za odobrenje proizvoda na tržištu da bi se prozivod bio komercijalno dostupan ili licenciran, npr.:
- FDA Biologics License Application (BLA) za biološke proizvode – zahtjev za dopuštenje da se biološki proizvodi predstave u međudržavnom trgovanju i FDA New Drug Application (NDA) za komercijalizaciju malih molekula.
- EMA Marketing Authorization Applications (MAA) – jednom odobrena od strane Europske Komisije, centralno tržišno odobrenje vrijedi u svim državama članicama Europske Unije (EU), Islandu, Norveškoj i Lihtenštajnu.
Za objavu rezultata istraživanja u medicinskom časopisu, normativi znani kao Consolidated Standards of Reporting Trials (CONSORT) se obično koriste, da bi izvještaj u članku bio prezentiran na standardiziran i svrsishodan način.
Faze kliničkih istraživanja
Postoje tri glavne faze kliničkih istraživanja: Faza I-III. Faza IV predstavlja studije koje se provode nakon što je proizvod odobren na tržistu/u državi i ima široku primjenu. Takvim studijama se istražuje dodatna sigurnost i učinkovitost proizvoda u stvarnom svijetu i pruža uvid u dodatne informacije koje pristižu iz velikog uzorka (Figura 1).
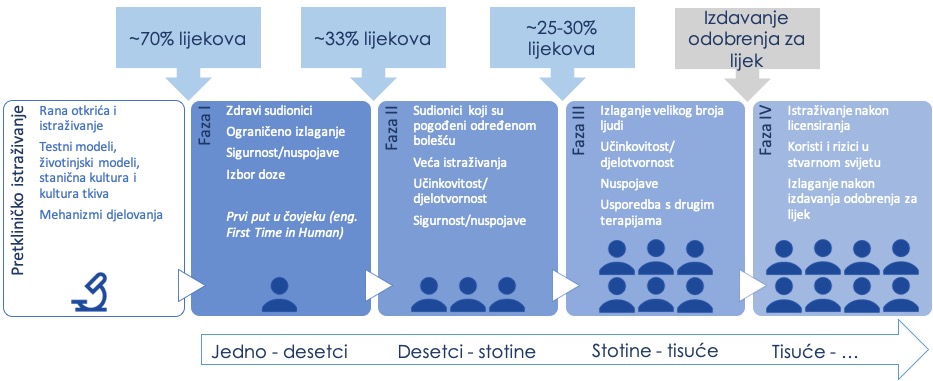
Faza I: je li sigurno?
Faza I studije su najmanje u svom opsegu i obuhvaćaju nekolicinu sudionika. Provode se u zdravim sudionicima, i zbog toga se koriste kako bi se pružio uvid u najčešće nuspojave u maloj populaciji. Također se zovu studijama “prvi put u čovjeku” (engl. “First Time in Human” (FTiH)) jer se intervencija daje ljudima po prvi put ikada. Glavni cilj takvih studija je ustanoviti sigurnost prozivoda koji se koristi. Obično traju nekoliko mjeseci do godine dana.
Faza II: da li funkcionira?
Faza II studije se primjenjuju da se dodatno istraži sigurnost proizvoda (rizici, nuspojave) u ciljnoj populaciji (npr. u pacijentima s određenom bolesti/stanjem ili trudnicama) u manjem, kraćem opsegu u usporedbi s Fazom III. Veličina uzorka je veća u odnosu na Fazu I i takve studije mogu služiti kao daljnje studije traženja doze (engl. dose-finding studies; pronalazak odgovarajuće doze proizvoda) i raznih režima (koja učestalost primjene terapije je najprikladnija). One pružaju uvid u učinkovitost lijeka iako to nije njihov glavni cilj. Faza II studije traju obično oko 2 godine.
Faza III: da li je učinkovito?
Faza II studije su u suprotnosti s Faza III studijama koje dulje traju, veće su u opsegu i koje ispituju učinkovitost/djelotvornost lijeka za određenu bolest u ciljnoj populaciji. Koriste se također da se ustanovi je li novi lijek bolji od onoga koji se trenutno koristi na tržištu. Faza III može trajati nekoliko godina i znači veliku vremensku i novčanu investiciju.
Nije rijetko da se studije spajaju u Faza I/II ili II/III dizajne u istoj studiji koja je podijeljena na dijelove: npr. Dio I jedne studije odgovara Fazi I i Dio II odgovara Fazi II iste studije. Takve studije se nazivaju bešavnim (engl. seamless) i prilagodljivim jer spajaju više faza unutar jedne studije. Završna analiza takve studije bi sadržavala rezultate svih sudionika koji su primili proizvod i komparator/placebo (ukoliko je korišten) u oba dijela (npr. Faza I i Faza II). Prilagodljiv pristup znači da ste studija može planski prilagoditi tijekom vremena, ovisno o rezultatima privremene analize na nemaskiranim podatcima.
Dizajn kliničkih istraživanja
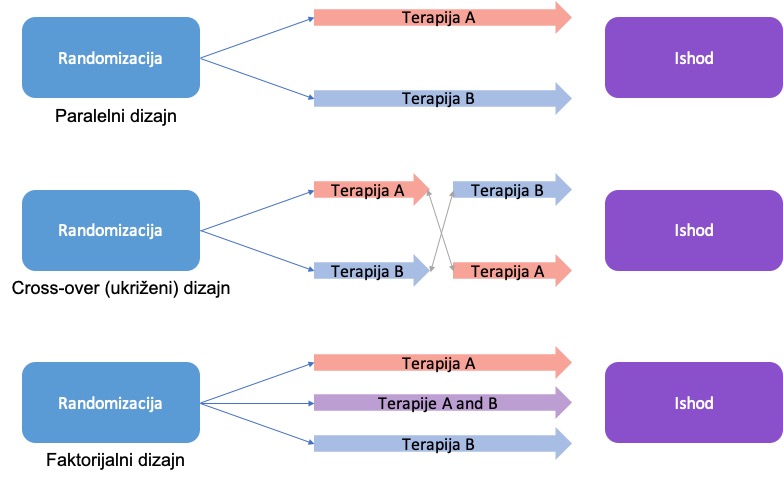
Osim faza kojima pripadaju, studije je moguće dizajnirati na različite načine. Najjednostavniji dizajn kliničke studije je paralelni dizajn (engl. parallel design). Takva studija podrazumijeva više grupa koje se istražuju i koje odgovaraju 1 ili više doza novog proizvoda i placebo/komparator grupi (ukoliko se koristi), prilikom čega je početak za sve grupe istodoban. Paralelni dizajn podrazumijeva velik uzorak kako bi se jasno mogle razlučiti razlike u učinku između više zadanih grupa sa statističkom sigurnošću. To je možda najkraći put za odabir prikladne finalne doze jer se sve terapije daju jednako učestalo tijekom cijele studije. Paralelni dizajn može biti i sekvencionalan = proveden serijskim redoslijedom, počevši s najnižom dozom i nastavljajući sa svakom novom, višom dozom, kada je ustanovljena sigurnost prethodne doze (eskalacija doze). Takva studija bi trajala dulje u odnosu na nesekvencionalan paralelni dizajn, jer svaki korak mora proći zasebnu procjenu prije nego novi započne.
Faktorijalni dizajn (engl. factorial design) je prikladan za studije koje se bave jednom dozom i uključuje 2 ili više tretmana dana istovremeno. Najjednostavniji faktorijalni dizaj je 2×2 dizaj, prikazan u Tablici 2 na primjeru rasta biljke – kako rast biljke ovisi o količini svjetla i vode koje biljke istovremeno primaju. Taj primjer se može primijeniti i na kliničke studije. Ukoliko su grupe uravnotežene u smislu veličine uzorka i osnovnih osobina, faktorijalnim dizajnom se može proučavati zajednički učinak više lijekova.
| Učestalost zalijevanja | |||
| Dnevno | Tjedno | ||
| Izlaganje suncu | Visoko | Rast biljke | Rast biljke |
| Nisko | Rast biljke | Rast biljke |
Sljedeći primjer često korištenog dizajna u testiranju novih lijekova je ukriženi dizajn (engl. crossover design), gdje se različite kombinacije lijekova mogu davati tijekom određenog vremena, nakon čega slijedi period čišćenja (engl. washout = period bez primanja ikakvog lijeka) i zatim se daje druga doza/drugi lijek s istom učestalošću i trajanjem kao prva doza/lijek istoj osobi. Tako, varijabilnost između reakcija je smanjena jer ista osoba prima obje terapije u različitom vremenu. Ovaj pristup se ne može koristiti u istraživanjima cjepiva jer ne postoji primjeren period čišćenja.
Dodatni dizajni koji se upotrebljavaju u studijama također postoje (npr. klaster-randomizirane, , košara-, kišobran- i stepenasti klin-studije – engl. cluster randomised, basket and stepped wedge trials) ali u najvećem broju slučajeva se koriste tri vrste studija opisane u tekstu gore i prikazane na Figuri 2.
Bez obzira koji je dizajn odabran, studija može proučavati superiornost novog lijeka naspram drugog ili placeba, neinferiornost naspram drugom lijeku ili ekvivalentnost u smislu djelotvornosti:
Studije superiornosti služe kako bi se pokazalo da je nova terapija bolja od dostupnog proizvoda ili placeba. Kada nema odgovarajuće alternative, samo novi proizvod, tada je uvijek cilj studije pokazati supriornost tog novog proizvoda.
Studije neinferiornosti služe kako bi se pokazalo da je nova terapija barem dobra kao ona dostupna. Npr., može se dogoditi da je novi lijek jeftiniji ili manje štetan od starog lijeka i dodatno želimo istražiti da je djelotvoran kao i stari lijek. Studije neinferiornosti se također mogu koristiti prilikom kombiniranja cjepiva: istovremeno cijepljenje s 2 cjepiva mora biti jednako dobro (djelotvorno i ne više štetno) kao kad se dva cjepiva daju odvojeno.
Studije (bio)ekvivalencije služe kako bi se pokazalo da je novi lijek jednak starom lijeku. Ovake studije se obično koriste prilikom registracije generičkih lijekova jer generici moraju imati jednako djelovanje kao i brendirani lijek.
Složenost u provođenju kliničkih istraživanja
U prosjeku, vrijeme razvoja lijekova je oko 12 godina do odobrenja za širu upotrebu i proces koji najdulje traje i koji je ujedno i najskuplji je kliničko istraživanje. Od sveukupnog troška razvoja proizvoda, klinička istraživanja zauzimaju 60% troškova. Vrijeme i investicija se razlikuje ovisno o tome koliko je teško razviti određeni proizvod i o tome koliko žurno proizvod mora biti dostupan na tržištu, ali klinička istraživanja su općenito strahovito kompleksna, dugotrajna i iziskuju velik dio napora. Prema više izvora, samo oko 10-15% lijekova/cjepiva koja se istražuju u kliničkim studijama bivaju odobrena za široku uporabu od strane regulatornih tijela.
Ipak, znanost napreduje svakim danom i iskustvo i znanje iz prethodnih studija koje su se bavile proučavanjem istih ili sličnih tehnologija nam pomažu da postignemo brži razvoj. Najveći primjer je razvoj COVID-19 cjepiva koji pokazuje da se cjepivo bez presedana može razviti u vremenima bez presedana, otvarajući prostor za budućnost kliničkih studija.
Kako bi naučili više o specifičnostima kliničkih studija koja se bave istraživanjem cjepiva i zašto su znanstvenici bili u mogućnosti izbaciti COVID-19 cjepiva na tržište tako brzo, očekujte budući članak “Klinička istraživanja – treći dio: “Cjepiva u kliničkim istraživanjima”)”.
Bibliografija
Committee on Strategies for Responsible Sharing of Clinical Trial Data; Board on Health Sciences Policy; Institute of Medicine.Washington (DC): “Sharing Clinical Trial Data: Maximizing Benefits, Minimizing Risk.” National Academies Press (US); 2015 Apr 20.
Day S. J. and Altman D. G. Blinding in clinical trials and other studies, BMJ 2000; 321:504.
AB Tasty. 2022. “Statistics: What are Type 1 and Type 2 Errors?.” Last edited on 22 September 2018. https://www.abtasty.com/blog/type-1-and-type-2-errors/
American Cancer Society. 2022. “Types and Phases of Clinical Trials.” Last edited on 18 August 2020. https://www.cancer.org/treatment/treatments-and-side-effects/clinical-trials/what-you-need-to-know/phases-of-clinical-trials.html
Basicmedical Key. 2022. “Trial Design: Overview of Study Designs (Phase I, II, III, IV, Factorial Design).” Last edited on 26 September 2017. https://basicmedicalkey.com/trial-design-overview-of-study-designs-phase-i-ii-iii-iv-factorial-design/
Certara (by Nathan Teuscher). 2022. “Trial Designs—Non-inferiority vs. Superiority vs. Equivalence.” Last edited on 1 January 2011. https://www.certara.com/knowledge-base/trial-designs-non-inferiority-vs-superiority-vs-equivalence/
European Federation of Pharmaceutical Industries and Associations (EFPIA). 2022. “Clinical Trials.” https://www.efpia.eu/about-medicines/development-of-medicines/regulations-safety-supply/clinical-trials/
Lupus Research Alliance. 2022. “On Biostatistics and Clinical Trials.” Last edited on 1 August 2019. https://lupustrials.org/about-trials/phases-of-a-trial/
PennState Eberly College of Science. 2022. “Stat 509 – Design and Analysis of Clinical Trials.” https://online.stat.psu.edu/stat509/lesson/5
Pubrica Academy. 2022. “Phases of a Clinical Trial.” Last edited on 4 February 2019. https://pubrica.com/academy/statistical/on-biostatistics-and-clinical-trials/
Hoffmann-La Roche Ltd. 2022. “What is a clinical trial and how does a trial work?.” https://www.roche.com/research_and_development/who_we_are_how_we_work/research_and_clinical_trials/what_is_a_clinical_trial.htm
Scribbr (by Lauren Thomas). 2022. “Independent and Dependent Variables | Uses & Examples.” Last edited on 27 August 2021. https://www.scribbr.com/methodology/independent-and-dependent-variables/
Statology. 2022. “A Complete Guide: The 2×2 Factorial Design.” Last edited on 13 May 2021. https://www.statology.org/2×2-factorial-design/
Surveillance, Epidemiology and End Results (SEER) Program Training,. 2022. “Blinded Clinical Trial.” https://training.seer.cancer.gov/treatment/other/trial.html
Wikipedia. 2022 “Confounding.” Last edited on 2 January 2022. https://en.wikipedia.org/wiki/Confounding
Wikipedia. 2022 “Minimization (clinical trials).” Last edited on 4 April 2020. https://en.wikipedia.org/wiki/Minimisation_(clinical_trials)
Wikipedia. 2022 “Null hypothesis.” Last edited on 5 January 2022. https://en.wikipedia.org/wiki/Null_hypothesis